利用芳香族羧酸酐的各种合成方法
- 2013-01-11
- 专题
概论
脱水缩合试剂,室温下稳定,易处理,且反应性高,遵循这样的结果,人们发现,芳香族羧酸酐可以用作各种功能强大的试剂。通过使用TFBA或者BTFBA与路易斯酸催化剂反应可以为碳氧键的形成提供一个最有效的支持,使得所需的羧酸酯和内酯的合成有很好的收益率。此外,碱性条件下,MNBA或者DMNBA与亲核催化剂反应可以方便的制得酸敏感性羧酸酯、内酯和酰胺类。另外,利用PMBA或苯甲酸酐作为不对称催化剂可以实现外消旋仲醇与外消旋2-芳疾病算的动力学拆分,从而建了一个新的合成光学活性羧酸酯、仲醇、2-芳基丙酸的方法。
1. 介绍
有效地人工合成有机物质,就要求开发用于形成(1)碳碳键;(2)碳氢键;(3) 碳氧键;(4)高选择性的碳氮键的简便方法。随着有机合成二十多年来的飞速发展,多来越多的优秀的化学合成方法被开发出来。“无环立体控制合成方法”的建立,比如说形成区域选择性和立体选择性碳碳键的烯丙基化反应或者醇醛缩合反应使得近几年来构建分子量超过1000的有机分子碳骨架成为可能。对于碳氢键的形成,由于不对称氢化反应的不断进步等,易获得光学活性分子的绝对构型控制。
然而,与令人瞩目的(1)与(2)的研究相比较, (3)和(4)新反应形成的发展目前没有大的进展。在这种情况下,我们试图通过使用“芳香族羧酸酐” 作为缩合试剂来开发一系列新的碳-杂元素键的形成反应。由于这些反应在温和的条件下进行,并且没有有害物质比如重金属等的使用,因此这些方法合成有机分子对环境负担小,并且可以高选择性的在任何的位置形成碳杂元素键。在本文中,我们介绍一系列这些年中我们从开发到应用覆盖的研究成果。
2.利用“芳香族羧酸酐”的缩合反应的发现
在研究甲硅烷基烯醇醚化学性质的同时,我们也研究了羧酸的甲硅烷基酯并关注了其作为酰化试剂的效用。首先,我们发现了一个新的反应,其中路易斯酸催化剂的存在下,羧酸甲硅酯(亲电子试剂)与烷基甲硅醚(亲核试剂)之间发生一个亲核取代反应得到相应的羧酸酯,产率很高(Scheme 1)。1)

然而,这个反应是一个直接的将羧酸甲硅酯到羧酸酯的酯交换反应,并且需要甲硅烷氧基到烷氧基转换进程中的热量。
在此阶段,我们决定在温和的条件下使用不同的试剂来重新研发衣蛾制备羧酸酯的替代方法。
比如乙酰氯,苯甲酰氯常用于醇的酰化,乙酸酐也可用作适当的酰化试剂。另一方面,芳香族的羧酸酐反应性特别的差,所以他们很少用于醇的直接苯甲酰化。因此,我们假定苯甲酸酐作为酰基供体反应活性属性较低,相应的脂肪族羧酸酯可以从羧酸硅烷酯和烷基硅烷醚中选择性的获得(Scheme 2)。
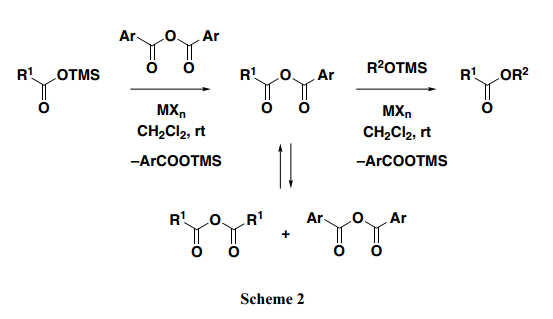
首先,酰基转换反应在脂肪族羧酸硅烷酯和芳香族羧酸酐之间平稳进行。接着,混合酸酐部分快速的生成歧化为两种类型的对称酸酐,因此,这三种酸酐整体上在反应系统中必须快速的进入平衡混合状态。由于通常情况下酸酐的反应性在对称芳香族羧酸酐<对称脂肪族羧酸酐<混合酸酐顺序中逐渐增高,所以可以假定最活跃的酸酐与亲核试剂优先反应。此外,如果混合酸酐中脂肪族酸酐部分的反应性高于芳香族酸酐部分的反应性,烷基甲硅烷基醚,必须优先攻击的脂族酰基部分,得到相应的脂族羧酸酯,同时伴随着芳族羧酸的甲硅烷基酯的消除。
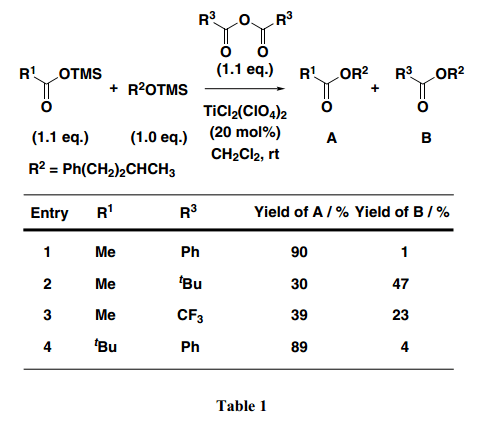
事实上,4-苯基-2-丁醇的三甲基硅烷醚加入预先准备好的混合酸酐中(使用TiCl2 (ClO4)2作为路易斯酸催化剂从乙酸三甲基硅烷酯和苯甲酸酐制得),得到所期望的酯A 90% 产率, 同时苯甲酸酐与三甲基硅烷基醚直接反应得到没有计划的酯B (1%产率)(Table 1, Entry 1)。结果显示,第一阶段的反应苯甲酸酐平稳进行转换为混合酸酐,苯甲酸酯的形成速率比脂肪族酸酯慢得多。当使用其他可用酸酐时,得到同样的实验进程,结果如Entries 2–4所描述。尽管新戊醇或者三氟乙酸酐在合成肽中适用于制备混合酸酐的有效试剂,但是它们不适合用于缩合反应中。例如,在反应中使用酸酐,相当数量的不是预期期望的酯B(新戊酯或三氟酯)作为副产物成为意想不到的产物(Entries 2和3)。另一方面,如Entry 4所示, 新戊酸三甲基硅烷基酯使用苯甲酸酐转换得到A,产率很好,结果显示通常情况在这个条件下新戊酸酯会优先于苯甲酸酯形成。
3. 使用4-三氟甲基苯甲酸酐的酯化反应(TFBA)2)
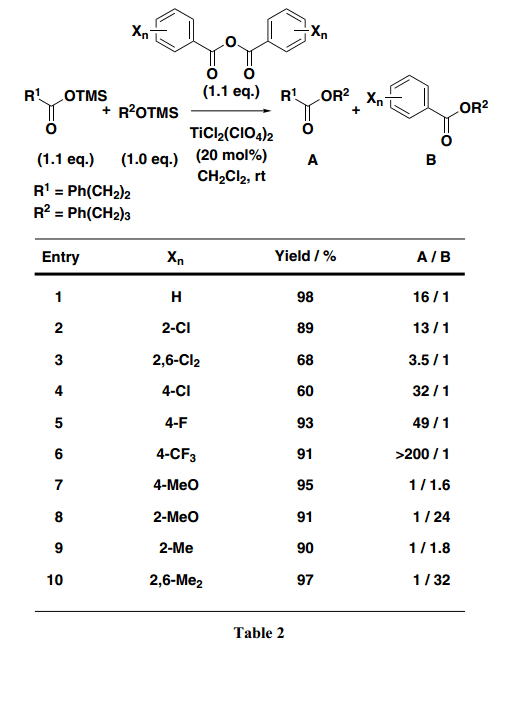
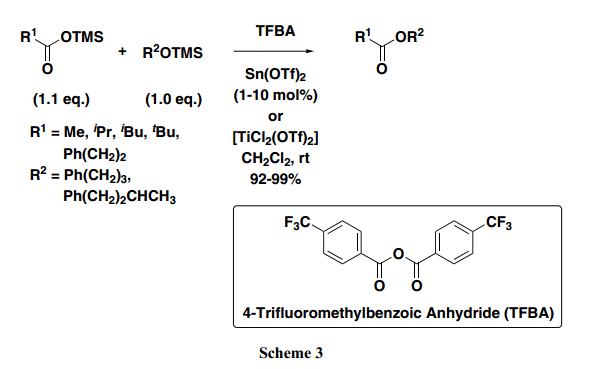
开发适合分子内缩合反应的合成方法,要求设计从几乎等量的两种起始原料中合成得到高产率目标产物。此外,为了提高环化产品的产率,很有必要开发一个新的合成具有完美选择性的酯的方法。因此,我们尝试在脱水缩合反应中在芳香环上引入多种取代基来增加其产率和选择性(Table 2)。例如,Entry 1与Entries 4–6 相比较显示出在芳香环4-位置处进入一个吸电子基团可以增加化学选择性。特别是,使用4-三氟甲基酸酐(TFBA)的反应得到选择性极好的预期产品脂肪族酸酯,并且通过NMR监测,几乎没有苯甲酸酯副产物生成。此外,如Entries 2和3所示,芳香环2-位置处引入氯产生相同或者略微弱些的选择性。另一方面, 在使用具有甲氧基或者甲基基团的取代苯甲酸酐条件下,选择性被逆转,优先得到不需要的苯甲酸酯,而不是预期想要的脂肪羧酸酯(Entries 7–10)。从这些结果可以发现,在芳香环4-位置处引入一个强吸电子基团十分有效;特别是,具有一个三氟甲基基团的TFBA作为一个缩合试剂功能非常好。接下来,我们路易斯酸仔细研究了酯化功能的促进,比如Sn(OTf)2和TiCl2(OTf)2。结果发现,异丁酸、异戊酸、新戊酸和3-苯基丙酸三甲基硅烷酯与烷基三甲基硅烷酯快速反应得到相应并且几乎等量的羧酸酯(Scheme 3)。此外,在TFBA存在下,酯化反应得到近90%或者更多的目标产物和不到约1 mol% 的路易斯酸。
作为一个酯化方法的应用案例,合成高纯度的共轭酸酯可得以实现。α,β-不饱和酯,比如巴豆酸、异戊烯酸和当硅酸酯在碱性条件下有进行异构化的可能性,这会造成产生副产物的麻烦。相反,在酯化反应中通过使用TFBA只有目标产物可以选择性的获得(Scheme 4)。2b)

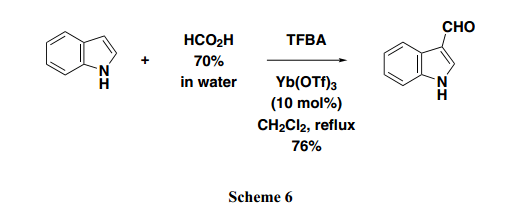
此外,应当指出,TFBA在合成活性酯中也十分有效,比如硝基苯酚酯和苯硫酚酯2b,3)取代基酰苯胺,4)和芳香酮5)(Scheme 5)。此外,这个方法也可有效的用于将甲酰基引入吲哚环中以及传统的方法比如Gattermann−Koch(加特曼-科赫反应)反应或者维尔斯迈尔反应中。因此,我们成功的开发了一个从甲酸直接制备3-甲酰基吲哚的有效方法(Scheme 6)。6)
4.利用TFBA的ω-甲硅氧烷基羧酸硅酸酯内酯化反应7)
将缩合方法适用于分子内反应有意想不到的难度。事实上,虽然有越来越多新和成方法的报道,但是目前有效的内酯合成还是很少。这是因为在反应设计中低浓度条件下提高分子内反应,以及开发新的试剂可以激活酯的羟基或羧基基团的选择性都很困难。为解决这些问题以合成天然产物的有效方法如下:(1) Mukaiyama−Corey method (S-pyridylester method);(2) Mukaiyama method (鎓盐法);(3)正宗方法(硫醇酯活化法);(4)Yamaguchi method(2,4,6-三氯苯甲酰氯/Et3N/DMAP方法);(5)Steglich−Keck method(DCC/DMAP·HCl method)。
然而,所有的反应通常都必须在高温该稀释条件下进行,使得分子内反应优于分子间反应。在这样的条件下,反应过程中使用不稳定的底物可以得到预期的环化产品,产率不好。我们尝试通过TFBA处理内酯化ω-羟基羧酸硅烷基衍生物与期望的羧酸部分选择性激活,从而导致所生成的混合酸酐快速的闭环。结果,化学选择性激活得以实现,显示最初由ω-羟基羧酸硅烷基酯和TFBA 所得的混合酸酐快速环化得到相应的大型内酯(Scheme 7)。这个方法使得制备13-元或更大型的内酯成为可能,并伴随有一些小量的相关26-元中型或大型内酯。此外,路易斯酸催化剂活性按Sn(OTf)2< TiCl2(SbF6)2< TiCl2(OTf)2< TiCl2(ClO4)2顺序依次增强。

5. 游离羧酸和醇通过使用TFBA和其类似物的缩合反应8)
在上诉反应中,要求必须预先从羧酸和醇中制备硅烷基衍生物。为了开发更为便利的方法,我们筛选了各类芳香酸酐作为有效的脱水试剂。虽然在开始时,有这样的问题,反应过程中一些催化剂失活导致产率差劲,我们最终通过使用比TFBA具有更强吸电子基环的3,5-双(三氟甲基)苯甲酸酐(BTFBA),得到了所需的羧酸酯,产率很好(Scheme 8)。
此外,通过使用3当量的三甲基氯硅烷,游离的羟基羧酸分子内脱水缩合得到所需的大环内酯,收益很好,类似于通过甲硅烷酯方法所得的情况。8b,9)在优化反应条件下,可使用市售TFBA代替内酯化。尤其是,12-羟基硬脂酸环化反应平稳进行,即使只有1mol%的路易斯酸条件下,得到产率为83%的13-元内酯(Scheme 9)。

此外,通过使用Group 4金属三氟甲磺酸根, Hf(OTf)4 作为路易斯酸,在TFBA的存在下平稳的极性宏酯化反应得到相应的内酯。另外,值得注意的是,中型环恩子也可以通过该方法得到。例如,通过采用这个方法Cephalosporolide D的8-元环内酯部分的合成得以成功的实现(Scheme 10)。10)

6. 使用2-甲基-6-硝基苯甲酸酐(MNBA)的酯合成11)
上述 “取代基苯甲酸酐方法”对制备耐酸性分子非常有效,然而,这在合成多官能团天然产物方面仍然有很多的结构性限制。因此,我们更为严密的筛选出新型的芳香族羧酸酐,碱性条件下可以通过亲核催化剂如DMAP 进行激活(Table 3)。如Entry 1所示,反应使用无取代苯甲酸酐得到目标脂肪族羧酸酯,中等产率,化学选择性好(A/B=100/1)。此外,当甲氧基基团引入到芳香环4-位置处时,化学选择性得到改善,然而,目标酯A 产率降低,因为反应抑制作用(Entry 2)。 此外,如Entries 3和4所示,芳香环4-位置处通过使用具有吸电子基的取代苯甲酸酐,例如TFBA,反应速率显著提高,然而,反应中也会有2–4% 产率的副产物取代基苯甲酸酯B生成。

考虑查到这样的结果,我们在芳香环2-位置处对取代基效果做了进一步的检测,期望羰基周围的位阻影响可以改进化学选择性。在Entries 5–7,我们尝试通过使用2-位置或者2-和6-位置具有吸电子基团甲基或者甲氧基的取代基苯甲酸酐合成羧酸酯。这些反应整体上显示出良好的选择性,将副产物B产率降至1%或者更少。然而,由于反应性降低,这个条件的结果对内酯化仍然不足。两外, Entries 8和9所示,反应中使用2-和6-位置处都具有氯的芳香酸酐,混合酸酐生成,酯化反应顺利进行,然而,与使用TFBA相比,化学选择性没有得到改善。这些结果表明方向换上有吸电子基团对于高反应速率非常重要,另一方面 芳香环2-位置处引入给电子集团会出现高的化学选择性。基于这些现象的考虑,我们设计了2-甲基-6-硝基苯甲酸酐(MNBA),希望通过利用它改善反应性和化学选择性来足以的应用于分子内的反应。当我们使用MNBA(由2-甲基-6-苯甲酸)时,在当前的反应模板中,人们发现,所需的缩合反应快速进行,并且没有副产物2-甲基-6-硝基苯甲酸酯的生成(Entry 10)。
我们一直在不断的研究天然产物的合成,使用各种基材利用MNBA 方法极性分子内和分子间的缩合反应。在很多情况下,所需产物的合成产率会高于其他很多现有的合成方法,基质的一般性验证了我们的方法。如下图所示,利用MNBA成功的实现antibacterial Botcinic Acid的全合成(Scheme 11)。12)

7. 使用MNBA和其类似物的内酯化反应11b,13)
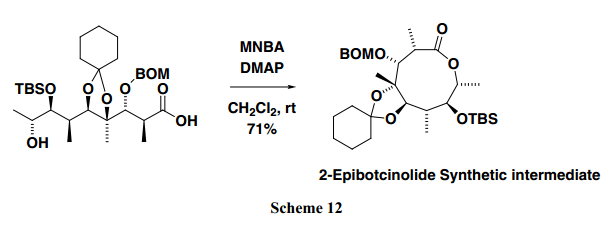
自从我们发现了使用MNBA的有效分子间缩合的最佳条件,我们也采用这个方法来分子内缩合。反应过程只需要将ω –羟基羧酸溶液室温下通过注射泵缓慢加入MNBA和DMAP溶液中(二氯甲烷或甲苯作溶剂)。在这些反应调价下,混合酸酐生成逐渐进行,并得到相应的内酯。因此,反应过程中MA的浓度始终很低,这种高稀释条件下提高了单体选择性。此外,MA激活可以在室温下完成,代替回流条件,这是一个简单的程序,通过亲核试剂阻止基料的降解 。值得注意的是我们成功的合成了2-Epibotcinolide的9-元内酯部分,首先假定Botcinin E化合物是通过应用MNBA方法而得(Scheme 12)。14)
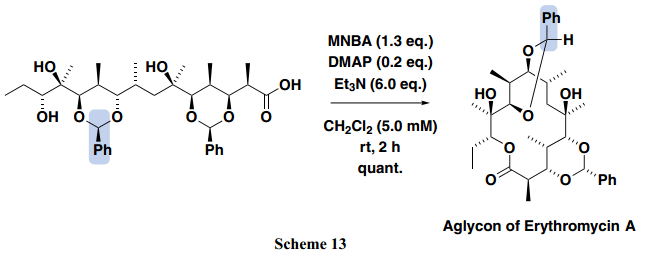
当MNBA方法被用于制备红霉素A苷元时,定量的产率得到所需的14-元内酯。15) 根据以往的红霉素A苷元合成的报道,知道环化效率不同源于seco acid 引入的前体保护基团。如Scheme 13所示,seco acid的内酯化,它具有一个适合闭环的结构,室温下段时间内能够快速的进行生成定量的红霉素A苷元。值得注意的是,即使是20 mol%的DMAP活化剂,也足够用来内酯化。具有类似于seco acid结构的原料的环化反应一直以来被认为得不到所需的内酯,然而,使用MNBA加热条件下的内酯化取得了令人满意的所需内酯。
这个方法更适合用于大型内酯的合成,例如,一个seco-acid 呗成功的转换为相应的25-元α-氧代内酯。这个中间酯最后脱保护得到2-hydroxytetracosanolide,昆虫白蚁的防御性唾液分泌(Scheme 15)。16)

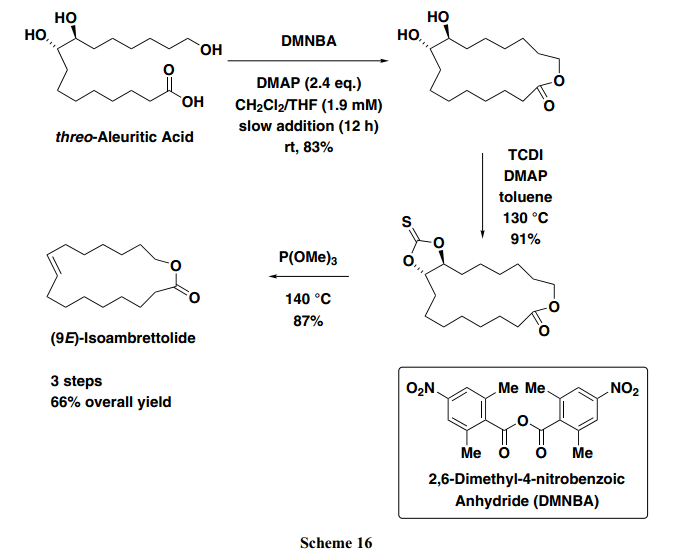
此外,筛选出具有各种吸电子基芳香酸酐之后,人们发现,通过使用2,6-二甲基-4-硝基苯甲酸酐(DMNBA),其2-和6-位置具有甲基,并且4-位置处具有一个强吸电子基,可以得到相应的羧酸酯和内酯,产率很好,并且相当于使用MNBA的量。由于使用MNBA 和DMNBA 的反应通常情况下在温和条件下进行,具有多个羟基的达到区域选择性seco-acid成功环化。如下图所示,人造麝香化合物 (9E)-异黄葵内酯在三个步骤中从非保护的油桐酸中成功制得,产率为66% (Scheme 16)。17)
8.具有活性优于DMAP 的吡啶N-氧催化剂DMAPO的发展
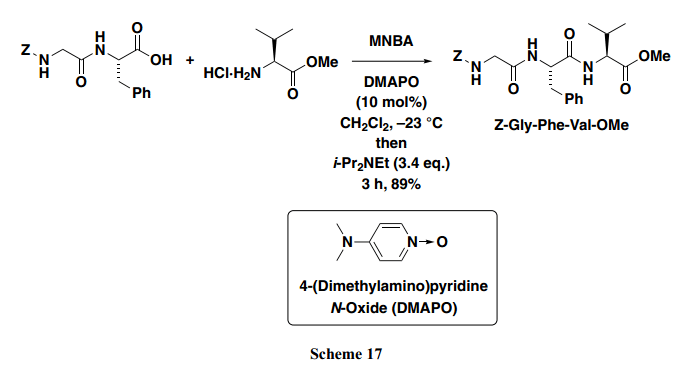
MNBA和DMAP的组合使得从羧酸和胺中合成羧酸酰胺成为可能。然而,这个方法中肽偶合的应用不合实际,由于其外消旋作用。碱性条件下筛选合适的催化剂交替DMAP后,发现4-二甲基氨基吡啶N-氧化物(DMAPO),已被用于有机磷酸酯类化合物的合成,可以有效地应用于肽偶合。例如,Z-Gly-Phe与Val-OMe 反应得到相应的肽,并没有外消旋作用(Scheme 17)。18)
有这些结果人们发现,通过使用芳香族羧酸酐作为缩合试剂,DMAPO是一个极好的内酯化催化剂。如下所示为Octalactin A,一个抗肿瘤海洋内酯的全合成, DMAPO(2–10 mol%)的催化量有效地使得8-元环部分形成,得到所需的中间体,产率高达90%(Scheme 18)。19)
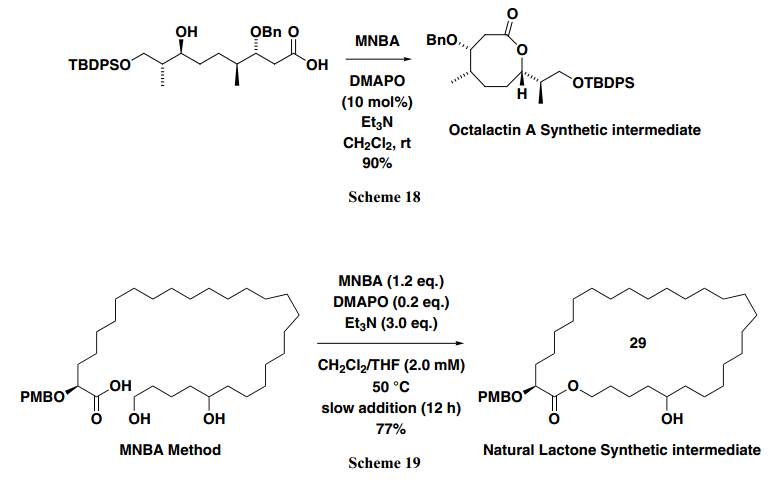
MNBA/DMAPO方法也可成功的引入大型宏内酯的形成(Scheme 19)。13b,16,20)如下所示,通过MNBA和DMAPO的结合成功的实现白蚁防御性唾液分泌的合成, 得到相应的单羟基内酯,产率高,区域选择性好。通常,seco acid,具有较少取代基长,趋于稳定的线性结构防止内酯形成。因此,通过我们的方法可以成功的获得所需的大环内酯,产率极好(77%)。另一方面,当我们与Yamaguchi method 作比较时,后者产率仅为29% (Scheme 20)。

9.使用芳香族酸酐作为缩合剂的不对称酯化反应的发展21-23)
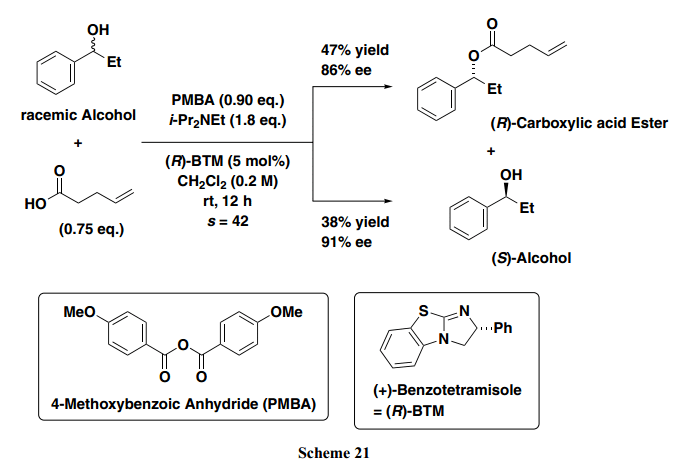
前面的章节中,非手性亲核试剂用作促进剂。考虑到在不对称合成中进一步的应用,我们决定筛选出手性亲核试剂代替DMAP。由Birman等人推广的(+)-苯并四咪唑 [(R)-BTM]24) 是一个很好的不对称催化剂可以满足我们的目的需求。通过和我们的脱水耦合方法相结合,对应选择性酯化可以实现。事实上,在4-甲氧基苯甲酸酐(PMBA)或苯甲酸酐的存在下, 外消旋二级苄醇与非手性羧酸反应得到过量的相应的(R)-羧酸酯和(S)-仲醇对映体(Scheme 21)。21)
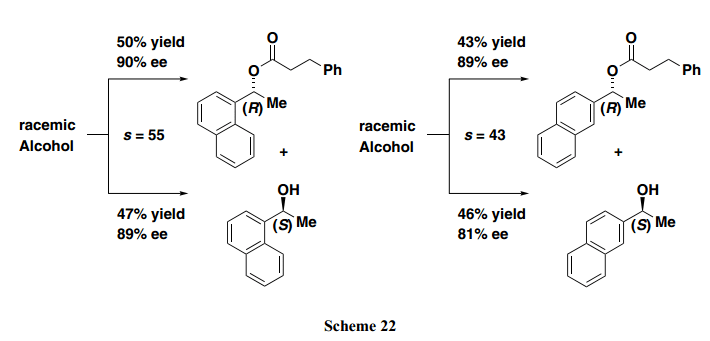
1-(α-萘基)-1-乙醇和1-(β-萘基)-1-乙醇与(R)-BTM和 苯甲酸酐的动力学拆分可以成功的完成,具有良好的s –值,分别为55和43(Scheme 22)。这里的s 值是表示两种对映体之间反应速率比的一个因素。通常,如果s值超过20,动力学拆分实际上是有用的。例如,在外消旋1-(α-萘基)-1-乙醇的反应中,(R)-酯成功获得,产率为50%(90% ee)。此外,剩余的(S)-醇也可以47%产率回收(89% ee)。
通过这种新颖的不对称催化,很有可能建立一个外消旋羧酸动力学拆分的新的方法。筛选适合这个目标的非手性醇之后,发现双(α-萘基)甲醇是一个非常好的亲核试剂。事实上,PMBA或苯甲酸的催在下,当外消旋2-芳基丙醇与双(α-萘基)甲醇反应进行时,不对称脱水反应顺利进行,得到高产率高对映体的目标产物 (R)-羧酸酯,以及剩余的具有中等对映选择性的(S)-2-苯基丙酸(Scheme 23)。22)

具有2-芳基丙酸部分作为基础结构的化合物被广泛的用作非甾体类抗炎药 (NSAIDs)。我们还成功的建立了一个新的合成方法,该方法提供光学活性NSAIDs和高选择性的酯衍生物(Scheme 24)。22)此外,所得双(α-萘基)甲基酯常规条件下很容易氢化裂解,光学纯度没有任何损失。因此,所得的手性羧酸酯可以被认为是游离羧酸的前体。
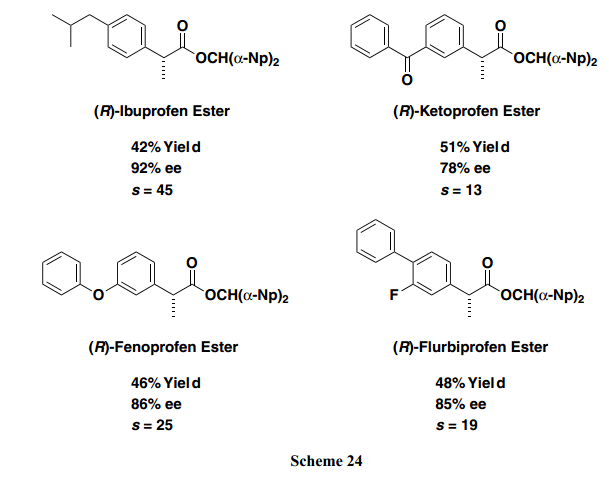
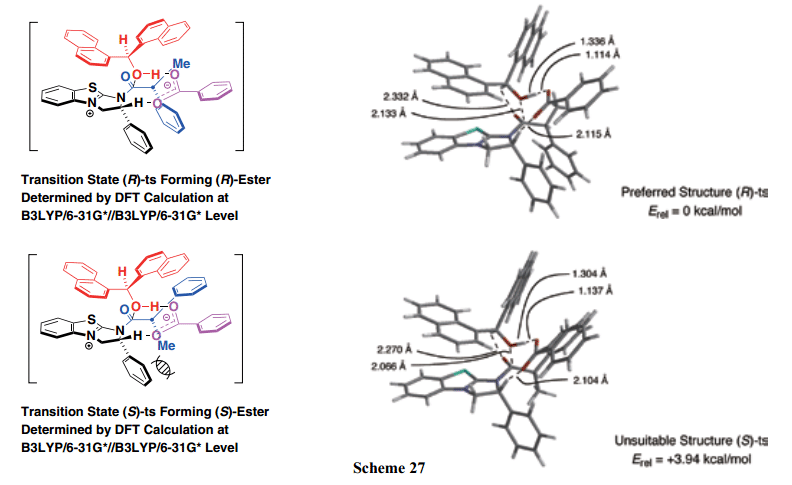
此外,我们成功的研发了(S)-β -Np-BTM, 一个新型亲核催化剂,优化了外消旋α –芳基丙酸的动力学拆分。(S)-β-Np-BTM的使用时的外消旋萘普生有效地动力学拆分,其中最终得到(S)-萘普生,一个高光学纯度抗炎物质(Scheme 25)。22)T外消旋羧酸动力学拆分的可能途径如下所示(Scheme 26):亲核催化剂存在下,苯甲酸酐和外消旋羧酸碱性条件下快速进行反式酰基化反应得到外消旋混合酸酐(MA),暂时形成一个关键的中间体。在这两个对映异构体中,混合酸酐通过(R)-BTM 优先激活从(R)-羧酸中生成[(R)-MA],然后,与双(α–萘基)甲醇反应选择性得到相应的(R)-羧酸酯。另一方面,由(S)-羧酸得来的混合酸酐[(S)-MA]仍旧没有反应。为了支持上述预测的机制,我们确定的优选的两个过渡态(R)-ts和(S)-ts((R)-和(S)-羧酸中衍生得来)分别通过使用密度泛函理论(DFT)在B3LYP/6-31G*//B3LYP/6-31G*水平计算。这些过渡态的能量比较表明(R)-ts的能量低于(S)-ts。如Scheme 27所示,由于(S)-ts的α –位置处的甲基取代部分和BTM C-2位置处的苯基基团之间的空间排斥,(S)-ts稳定性降低,并且所需的(R)-羧酸选择性获得。因此,我们通过使用计算化学方法已经公开了不对称缩合反应中enantio-discrimination的反应机制和起源。


10. 结论
总之,我们简要概述了我们实验室中所研究的脱水缩合反应的发展。阐明了芳香族羧酸酐(室温下稳定易处理的花歌舞)的功能,与足够的催化剂结合使用可以用作高反应性能的偶合试剂。TFBA 或BTFBA与酸性催化剂组合,或者MNBA 或DMNBA与亲核催化剂结合,碱性条件下适用于羧酸酯和内酯的合成。TFBA 和MNBA都有市售,建议您尝试一下。另一方面,DMAPO, 这个新型的催化剂也发挥了有效地作用。DMAPO水合物形式也有市售,可以用于比如升华的脱水过程。DMAPO催化能力高于DMAP,值得注意的是,通过使用DMAPO,各种肽类可以得到制备,并且可以没有明显的外消旋环化。因此,游离羧酸和醇不对称偶合中,PMBA或苯甲酸酐功能可以用作一个有效地脱水缩合试剂。23) 使用这些组合与不对称催化剂(R)-BTM或(S)-β-Np-BTM很容易的获得各种光学活性酯,羧酸和醇。
最后,我们将不胜感激更多的研究人员使用并评估该方法,我们希望我们的研究结果将有助于有机化学合成的发展。
References
1) I. Shiina, T. Mukaiyama, Chem. Lett. 1992, 2319-2320.
2) (a) T. Mukaiyama, I. Shiina, M. Miyashita, Chem. Lett. 1992, 625-628. (b) M. Miyashita, I. Shiina, S. Miyoshi, T. Mukaiyama, Bull. Chem. Soc. Jpn. 1993, 66 , 1516-1527.
3) T. Mukaiyama, M. Miyashita, I. Shiina, Chem. Lett. 1992, 1747-1750.
4) (a) M. Miyashita, I. Shiina, T. Mukaiyama, Chem. Lett. 1993, 1053-1054. (b) M. Miyashita, I. Shiina, T. Mukaiyama, Bull. Chem. Soc. Jpn. 1994, 67, 210-215.
5) (a) T. Mukaiyama, K. Suzuki, Chem. Lett. 1992, 1751-1754. (b) K.Suzuki,H. Kitagawa, T. Mukaiyama,Bull. Chem. Soc. Jpn. 1993, 66 , 3729-3734.
6) I. Shiina, M. Miyashita, M. Nagai, T. Mukaiyama, Heterocycles 1995, 40 , 141-148.
7) T. Mukaiyama, J. Izumi, M. Miyashita, I. Shiina, Chem. Lett. 1993, 907-910.
8) (a) I. Shiina, S. Miyoshi, M. Miyashita, T. Mukaiyama, Chem. Lett. 1994 , 515-518. (b) I. Shiina, Tetrahedron 2004 , 59, 1587-1599.
9) I. Shiina, T. Mukaiyama, Chem. Lett. 1994, 677-680.
10) (a) I. Shiina, Y. Fukuda, T. Ishii, H. Fujisawa, T. Mukaiyama, Chem. Lett. 1998, 831-832. (b) I. Shiina, H. Fujisawa, T. Ishii, Y. Fukuda, Heterocycles 2000 , 52 , 1105-1123.
11) (a) I. Shiina, R. Ibuka, M. Kubota, Chem. Lett. 2002, 286-287. (b) I. Shiina, M. Kubota, H. Oshiumi, M.Hashizume, J. Org. Chem. 2004, 69 , 1822-1830. See also, (c) I. Shiina, Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.), 2005, 63 , 2-17.
12) (a) H. Fukui, S. Hitomi, R. Suzuki, T. Ikeda, Y. Umezaki, K. Tsuji, I. Shiina, Tetrahedron Lett. 2008, 49 , 6514-6517. [Cover Feature Article] See also, (b) H. Fukui, I. Shiina, Org. Lett. 2008, 10, 3153-3156. (c) H. Fukui, I. Shiina, Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.), 2009, 67 , 628-642.
13) (a) I. Shiina, M. Kubota, R. Ibuka, Tetrahedron Lett. 2002, 43, 7535-7539. See also, (b) I. Shiina, H. Fukui, A. Sasaki, Nat. Protoc. 2007, 2, 2312-2317. (c) I. Shiina, Chem. Rev. 2007, 107, 239-273 .
14) (a) I. Shiina, Y. Takasuna, R. Suzuki, H. Oshiumi, Y. Komiyama, S. Hitomi, H. Fukui, Org. Lett. 2006, 8, 5279-5282. (b) I. Shiina, H. Fukui, Chem. Commun. 2009, 385-400. [Feature Article]
15) I. Shiina, T. Katoh, S. Nagai, M. Hashizume, Chem. Rec. 2009, 9, 305-320.
16) I. Shiina, A. Sasaki, T. Kikuchi, H. Fukui, Chem. Asian J. 2008, 3, 462-472.
17) (a) I. Shiina, R. Miyao, Heterocycles 2008, 76 , 1313-1328. See also, (b) I. Shiina, M. Hashizume, Tetrahedron 2006, 62, 7934-7939.
18) (a)I. Shiina, H. Ushiyama, Y. Yamada,Y.Kawakita,K. Nakata,Chem. Asian J. 2008, 3, 454-461. See also, (b) I. Shiina, Y. Kawakita, Tetrahedron 2004,60 , 4729-4733.
19) (a) I. Shiina, H. Oshiumi, M. Hashizume, Y. Yamai, R. Ibuka, Tetrahedron Lett. 2004, 45 , 543-547. (b) I. Shiina, M. Hashizume, Y. Yamai, H. Oshiumi, T. Shimazaki, Y. Takasuna, R. Ibuka, Chem. Eur. J. 2005, 11 , 6601-6608.
20) I. Shiina, T. Kikuchi, A. Sasaki, Org. Lett. 2006, 8, 4955-4958.
21) I. Shiina, K. Nakata, Tetrahedron Lett. 2007, 48 , 8314-8317. [Cover Feature Article]
22) (a) I. Shiina, K. Nakata,Y.Onda, Eur.J.Org.Chem. 2008, 5887-5890. (b) I. Shiina, K. Nakata, K.Ono, Y. Onda,M.Itagaki, J. Am. Chem. Soc. 2010, 132, 11629-11641.
23) For the asymmetric esterification, bulky fatty-chain carboxylic anhydrides, such as pivalic anhydride, are also effective as dehydrating reagents. See: (a) I. Shiina, K. Nakata, M. Sugimoto, Y. Onda, T. Iizumi, K. Ono, Heterocycles 2009 , 77 , 801-810. (b) I. Shiina, K. Nakata, K. Ono, M. Sugimoto, A. Sekiguchi, Chem. Eur. J. 2010, 16 , 167-172.
24) (a) V. B. Birman, X. Li, Org. Lett. 2006, 8, 1351. (b) V. B. Birman, L. Guo, Org. Lett. 2006, 8, 4859.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号